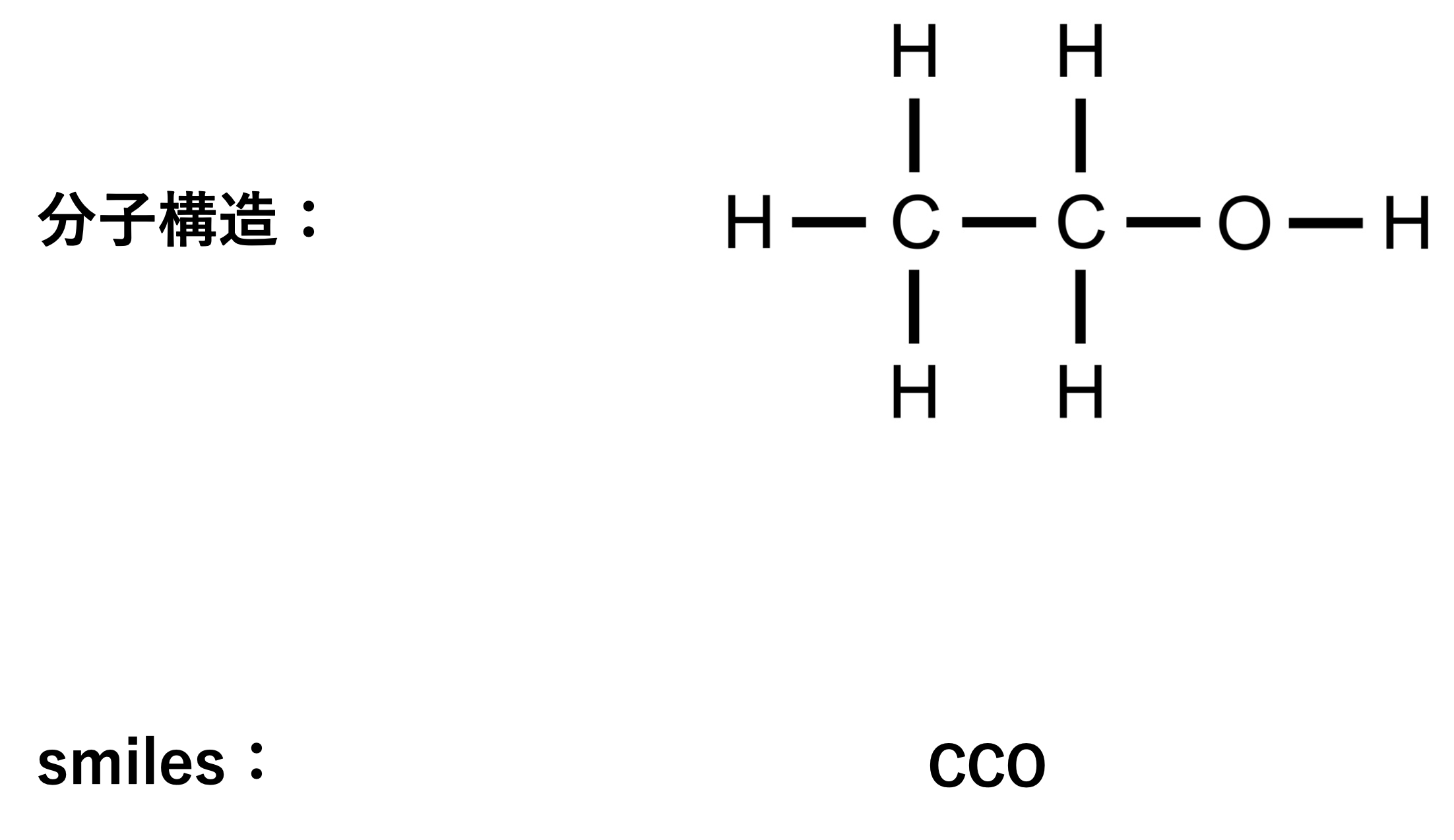
分子記述子として得られた多様な情報を活用し、機械学習の入力情報とすることで、非線形な関係や複雑な相互作用の学習が可能となります。溶解度パラメータを含む各種物性を機械学習で推定する場合、具体的には以下の流れになります。
1.分子構造(SMILESなど)から分子記述子を計算2.分子記述子と溶解度パラメータ(実測または先行研究データ)のデータセットを用意3.回帰モデル(ランダムフォレストなど)を学習4.新規分子構造の溶解度パラメータを推定
なお、機械学習モデルは様々な種類がありますが、溶解度パラメータのような連続値を扱うには回帰モデルが中心となります。
ランダムフォレスト:データ量が比較的少なくても安定した推定精度を得やすいサポートベクター回帰:適切なカーネル選択で高精度が見込めるニューラルネットワーク:大規模データや複雑な表現力が必要な場合に強みを発揮
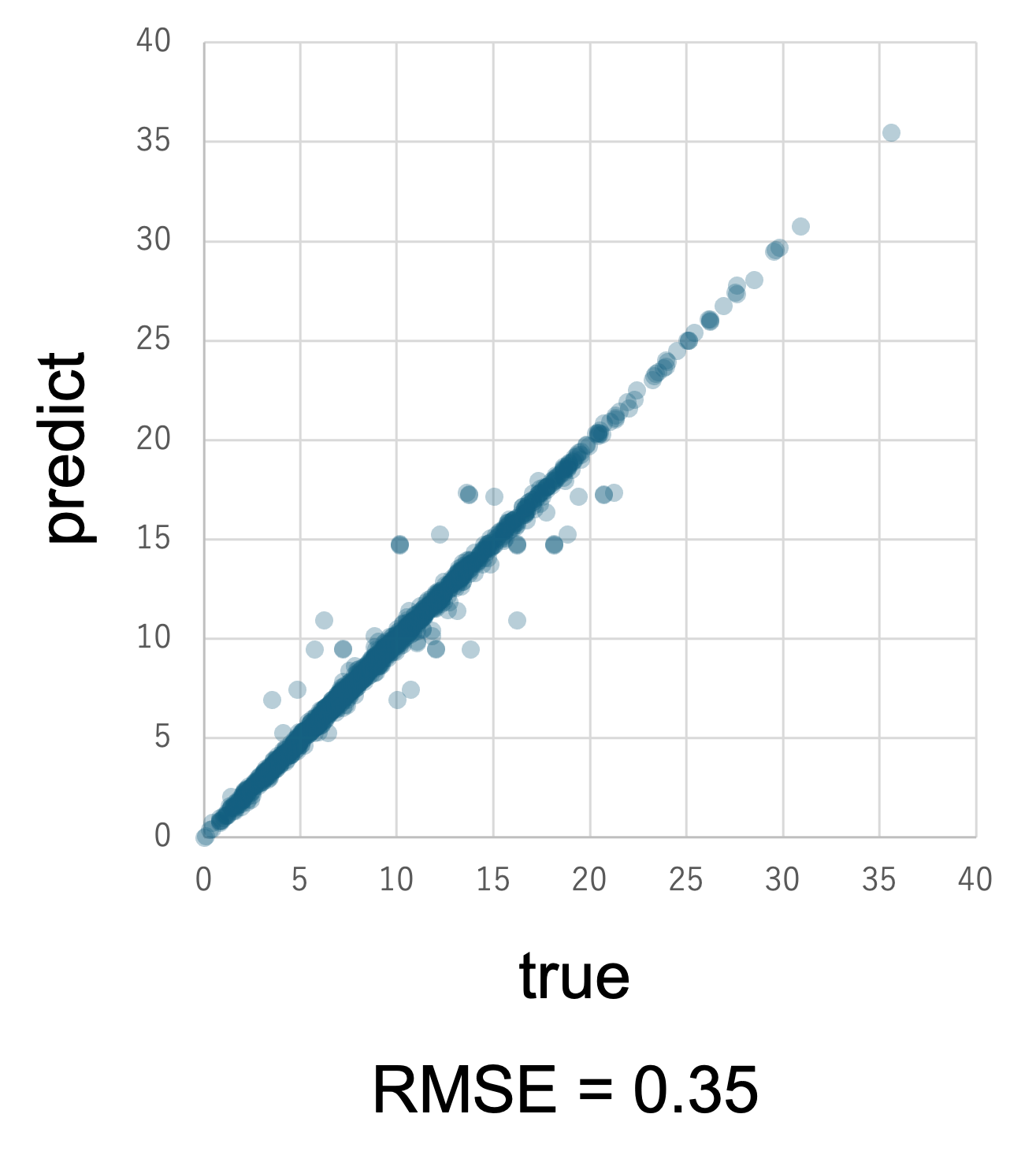
機械学習で得られたモデル性能は、決定係数R2やRMSE(Root Mean Squared Error)などの指標にて評価します。推定結果と実験値を比較しながらモデルをチューニングを行い、汎用性を高めていくことになります。
機械学習の拡張性
機械学習を用いた溶解度パラメータ推定の魅力は、その拡張性にあります。新たな実験データや分子構造情報が蓄積されることで、既存モデルに対して学習データの追加・更新が可能です。これにより、以下のようなメリットが期待されます。
対応領域の拡大:より広範囲な分子に適用可能となり、新規の分子構造への対応も可能になります。精度の向上:より細かい構造的特徴や物性の相関を学習でき、推定精度が更に向上します。シミュレーションとの統合:分子動力学シミュレーションや量子化学計算と組み合わせることで、理論的予測と実験結果の相補的な利用が可能となり、材料設計やデバイス開発の効率化に寄与します。
データベースの蓄積により、機械学習を活用した溶解度パラメータの推定の信頼性と適用範囲はさらに広がると予想されます。これにより、実験前のシミュレーションや設計段階での迅速な評価が可能となり、材料開発のスピードアップやコスト削減に大きく寄与することが期待されます。